Overview
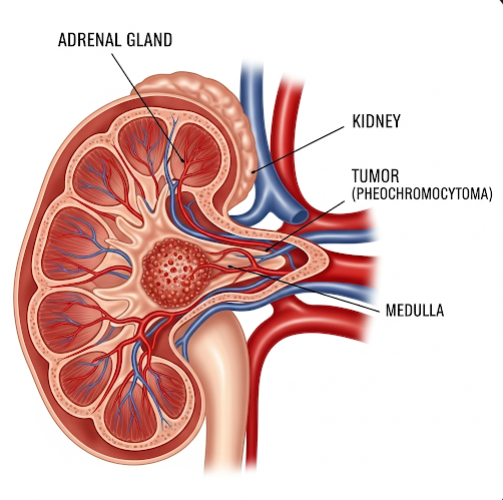
Pheochromocytoma is a rare, usually noncancerous tumor that forms in the adrenal glands, which are located on top of the kidneys. These tumors produce excessive amounts of catecholamines, particularly adrenaline (epinephrine) and noradrenaline (norepinephrine), hormones that control heart rate, blood pressure, and stress responses. The overproduction of these hormones can lead to episodes of dangerously high blood pressure and other life-threatening symptoms if left untreated.
What is Pheochromocytoma?
A pheochromocytoma originates from the chromaffin cells in the adrenal medulla (the inner part of the adrenal gland). These tumors may develop in one or both adrenal glands. In some cases, similar tumors—called paragangliomas—can occur outside the adrenal glands along the sympathetic nervous system.
Pheochromocytomas are typically benign, but around 10–15% can be malignant and spread to other parts of the body. Despite being rare, this condition is potentially curable with early diagnosis and surgical removal.
Symptoms
Symptoms of pheochromocytoma are usually episodic but can become chronic. They are caused by the sudden release of excessive catecholamines into the bloodstream:
- Severe, episodic high blood pressure (hypertension)
- Pounding headaches
- Rapid heartbeat (tachycardia) or palpitations
- Excessive sweating (diaphoresis)
- Tremors or shaking
- Paleness or flushing
- Shortness of breath
- Panic attack-like symptoms
- Nausea or abdominal pain
- Weight loss (in prolonged cases)
Attacks may be triggered by physical exertion, stress, surgery, or certain medications.
Causes
Pheochromocytomas occur when chromaffin cells begin to grow abnormally and overproduce hormones. While many cases are sporadic (no known cause), around 25–30% are linked to hereditary genetic syndromes, such as:
- Multiple Endocrine Neoplasia type 2 (MEN 2)
- Von Hippel–Lindau disease (VHL)
- Neurofibromatosis type 1 (NF1)
- Hereditary paraganglioma-pheochromocytoma syndromes
Genetic testing is often recommended, especially in younger patients or those with bilateral tumors.
Risk Factors
Several factors can increase the risk of developing pheochromocytoma:
- Family history of pheochromocytoma or paraganglioma
- Inherited genetic syndromes
- Age: Most commonly diagnosed between 30 and 50 years old
- High levels of stress or trauma may trigger symptoms in those with tumors
- Certain medications (e.g., decongestants, stimulants) can provoke attacks in undiagnosed cases
Complications
If left untreated, pheochromocytomas can cause severe or life-threatening complications:
- Hypertensive crisis (sudden, extreme rise in blood pressure)
- Heart attack or stroke
- Heart failure due to prolonged high blood pressure
- Organ damage (kidneys, brain, eyes)
- Arrhythmias (irregular heartbeat)
- Shock or death from hormone surges
- Malignancy (in cases where the tumor is cancerous)
Surgical removal of the tumor often resolves symptoms and prevents complications.
Prevention
Since pheochromocytomas are often genetically driven or sporadic, there are no guaranteed prevention strategies. However, you can reduce risks and improve outcomes by:
- Undergoing genetic screening if you have a family history of related syndromes
- Monitoring blood pressure regularly, especially if you experience unexplained spikes
- Managing stress and avoiding triggers that elevate adrenaline levels
- Seeking early medical care if you develop symptoms like episodic hypertension, palpitations, or anxiety without clear cause
- Routine imaging or screening for those with hereditary risk
Treatment Options in Korea
South Korea offers advanced diagnostic and treatment options for pheochromocytoma, with expert care available at top endocrine and surgical centers.
Diagnosis in Korea Includes:
- Blood and urine tests: Measurement of catecholamines and metanephrines
- Imaging tests: CT scans, MRI, or MIBG scintigraphy to locate tumors
- Genetic testing: For patients with family history or bilateral tumors
Treatment Approaches in Korea:
- Preoperative Management:
- Use of alpha-blockers (e.g., phenoxybenzamine) to stabilize blood pressure
- Beta-blockers may be added after alpha-blockade to manage heart rate
- Careful fluid management and diet prior to surgery
- Surgical Removal:
- Laparoscopic adrenalectomy is the most common and minimally invasive approach
- Open surgery may be needed for larger or malignant tumors
- Tumor removal usually cures hypertension and resolves symptoms
- Postoperative Care and Monitoring:
- Follow-up with endocrinologists to monitor hormone levels
- Surveillance imaging to detect recurrence
- Genetic counseling for patients with hereditary forms
Hospitals Offering Pheochromocytoma Care in Korea:
Top hospitals like Samsung Medical Center, Seoul National University Hospital, Asan Medical Center, and Yonsei Severance Hospital provide full-spectrum diagnosis, surgery, and genetic evaluation with English-speaking services for international patients.