Overview
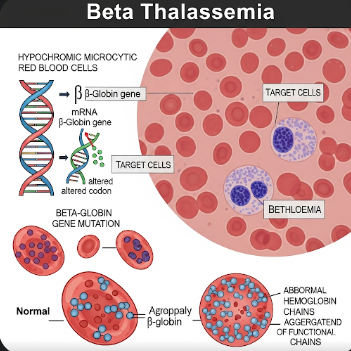
Beta Thalassemia is a genetic blood disorder characterized by reduced or absent production of beta-globin chains, an essential component of hemoglobin. This imbalance leads to anemia, ineffective red blood cell production, and in severe cases, organ damage due to iron overload. Although the condition is more common in Mediterranean, Middle Eastern, and Southeast Asian populations, it is increasingly recognized in Korea due to migration and improved genetic screening.
Modern Korean medical centers offer comprehensive care for beta thalassemia, including advanced diagnostic testing, transfusion therapy, iron chelation, and gene-focused interventions. Early diagnosis and long-term management are crucial to preventing severe complications and improving patient quality of life.
What is Beta Thalassemia?
Beta Thalassemia is an inherited disorder caused by mutations in the HBB gene, which encodes the beta-globin component of hemoglobin. Depending on the severity of the mutation, the disease manifests as:
- Beta Thalassemia Minor (Trait): Usually asymptomatic, carriers have mild anemia.
- Beta Thalassemia Intermedia: Moderate anemia requiring occasional transfusions.
- Beta Thalassemia Major (Cooley’s Anemia): Severe anemia requiring regular transfusions and intensive management.
The defective hemoglobin leads to ineffective red blood cell production in the bone marrow, causing chronic anemia, fatigue, and other systemic effects.
Symptoms
The clinical presentation varies by type:
Beta Thalassemia Minor:
- Mild fatigue or weakness
- Slight pallor
- Usually discovered incidentally during routine blood tests
Beta Thalassemia Intermedia:
- Moderate anemia with pallor and fatigue
- Delayed growth or puberty in children
- Occasional jaundice
- Bone deformities due to bone marrow expansion
Beta Thalassemia Major:
- Severe anemia from early infancy
- Marked fatigue and irritability
- Enlarged spleen and liver (hepatosplenomegaly)
- Bone deformities in the face and skull
- Jaundice and dark urine
- Growth retardation and delayed development
Causes
Beta Thalassemia is caused by inherited mutations in the HBB gene. Both parents must carry the defective gene for a child to develop Beta Thalassemia Major. Carriers (heterozygotes) usually have Beta Thalassemia Minor, which is generally asymptomatic. The severity of symptoms depends on the specific mutations inherited from each parent.
Risk Factors
- Family history of thalassemia or carriers of the HBB gene mutation
- Consanguineous marriages increase the risk of passing on severe forms
- Ethnic background (more common in Mediterranean, Middle Eastern, Southeast Asian, and certain Korean migrant populations)
Complications
Without proper management, Beta Thalassemia can lead to serious complications:
- Iron overload due to frequent blood transfusions, causing liver, heart, and endocrine damage
- Heart failure from chronic anemia and iron deposition
- Bone deformities and osteoporosis due to marrow expansion
- Delayed growth and puberty
- Infections due to splenomegaly or post-splenectomy status
- Liver disease or cirrhosis from iron accumulation
Prevention
Since Beta Thalassemia is genetic, prevention focuses on genetic counseling and early screening:
- Carrier testing for individuals with family history or at-risk ethnic backgrounds
- Prenatal testing for couples where both partners are carriers
- Avoiding consanguineous marriages in high-risk populations
- Early detection in infants to begin timely treatment
Treatment Options in Korea
Diagnosis
Korean medical centers use advanced testing for accurate diagnosis:
- Complete blood count (CBC) and hemoglobin electrophoresis
- Genetic testing to identify HBB mutations
- Prenatal diagnosis through chorionic villus sampling or amniocentesis in high-risk pregnancies
- Iron studies and imaging for monitoring iron overload
Medical Management
Blood transfusions are the cornerstone of treatment for moderate to severe cases:
- Regular transfusions maintain hemoglobin levels and prevent complications
- Iron chelation therapy (oral or intravenous) is essential to remove excess iron from repeated transfusions
Other supportive treatments include:
- Folic acid supplementation to support red blood cell production
- Monitoring for infections and organ function
- Vaccinations for patients with splenectomy or immunocompromised status
Advanced Therapies
Korean hospitals also offer modern interventions:
- Hematopoietic stem cell transplantation (HSCT): Potentially curative, especially for children with severe disease
- Gene therapy trials: Emerging therapies aimed at correcting the defective HBB gene
- Splenectomy for patients with severe splenomegaly or transfusion complications
Rehabilitation and Support
- Nutritional counseling to optimize iron management and general health
- Psychological support for chronic disease management
- Regular follow-up with hematology and endocrinology specialists
- Patient education on disease, transfusion schedules, and iron chelation adherence
Prognosis
With early diagnosis and comprehensive management, patients with Beta Thalassemia in Korea can achieve significantly improved life expectancy and quality of life. Regular transfusions, effective iron chelation, and access to advanced therapies like HSCT or gene therapy reduce the risk of severe complications. Long-term monitoring ensures that growth, organ function, and overall health are preserved.