Overview
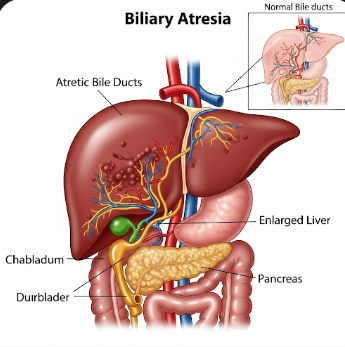
Biliary Atresia is a rare congenital liver disorder in which the bile ducts are either missing or blocked, preventing bile from draining from the liver to the intestines. This blockage causes bile accumulation in the liver, leading to liver damage, cirrhosis, and eventual liver failure if left untreated. Biliary Atresia is the most common cause of pediatric liver transplants worldwide.
In Korea, pediatric hepatology centers specialize in early diagnosis and surgical management of biliary atresia. Advanced imaging, timely surgical interventions, and comprehensive post-operative care contribute to improved survival rates and long-term outcomes for affected infants.
What is Biliary Atresia?
Biliary Atresia is a congenital obstruction of the extrahepatic and/or intrahepatic bile ducts, which carry bile from the liver to the gallbladder and intestines. Without bile flow, bilirubin builds up in the blood, causing jaundice and damaging liver cells.
The condition is classified into:
- Perinatal (acquired) form – develops shortly after birth, the most common type
- Embryonic (fetal) form – associated with other congenital anomalies
Left untreated, biliary atresia leads to progressive liver fibrosis, cirrhosis, and portal hypertension.
Symptoms
Symptoms usually appear within the first few weeks of life and may include:
- Jaundice (yellowing of skin and eyes) persisting beyond two weeks of age
- Dark urine due to bilirubin buildup
- Pale or clay-colored stools because of lack of bile in the intestines
- Enlarged liver or spleen (hepatosplenomegaly)
- Poor weight gain and growth retardation
- Irritability or discomfort in infants
- Advanced cases may show signs of ascites and liver dysfunction
Early recognition of these symptoms is critical, as timely surgical intervention can significantly improve prognosis.
Causes
The exact cause of biliary atresia remains unclear, but possible mechanisms include:
- Genetic factors – certain mutations may predispose infants to bile duct malformation
- Immune-mediated injury – abnormal immune responses may attack bile ducts postnatally
- Viral infections – some studies suggest that perinatal viral exposure may contribute
- Developmental anomalies during embryogenesis affecting bile duct formation
Risk Factors
- Female gender (slightly more common in girls than boys)
- Family history of biliary or liver anomalies (rare)
- Asian ethnicity shows a slightly higher incidence, including in Korea
- Premature birth or low birth weight may be associated with increased risk
Complications
If untreated, biliary atresia can result in:
- Progressive liver cirrhosis and chronic liver disease
- Portal hypertension, leading to varices and gastrointestinal bleeding
- Liver failure, requiring transplantation
- Growth retardation and malnutrition due to impaired fat absorption
- Increased risk of hepatocellular carcinoma in chronic liver disease
Prevention
Biliary Atresia cannot be fully prevented due to its congenital nature. However, early detection can improve outcomes:
- Newborn screening for prolonged jaundice
- Prompt medical evaluation for infants with pale stools or dark urine
- Genetic counseling in families with a history of biliary anomalies
Treatment Options in Korea
Diagnosis
Korean pediatric hepatology centers employ a structured approach for diagnosis:
- Physical examination for jaundice, hepatomegaly, and growth assessment
- Blood tests measuring bilirubin, liver enzymes, and clotting factors
- Abdominal ultrasound to evaluate bile ducts and liver structure
- Hepatobiliary scintigraphy (HIDA scan) to assess bile flow
- Liver biopsy to confirm bile duct injury and fibrosis
- Genetic testing in selected cases to identify syndromic associations
Surgical Management
Kasai Portoenterostomy is the standard surgical procedure:
- Performed as early as possible, ideally within the first 60 days of life
- Involves removing the damaged bile ducts and connecting the liver directly to the small intestine
- Restores bile flow and slows liver damage
For infants who do not respond to Kasai surgery or develop end-stage liver disease:
- Liver transplantation is performed
- Korea has advanced pediatric liver transplant programs with high survival rates
Medical Management
Post-surgery medical care focuses on supporting liver function and preventing complications:
- Ursodeoxycholic acid to improve bile flow
- Fat-soluble vitamin supplementation (A, D, E, K) due to malabsorption
- Antibiotics to prevent cholangitis, a common post-surgical complication
- Nutritional support to promote growth and weight gain
Rehabilitation and Support
- Regular follow-up with pediatric hepatologists for liver function monitoring
- Physiotherapy and growth monitoring
- Psychological support for families navigating chronic illness and transplantation
- Patient education on recognizing complications and maintaining optimal nutrition
Prognosis
The prognosis for biliary atresia depends on age at diagnosis and surgical intervention:
- Early Kasai surgery significantly increases survival and delays the need for transplantation
- With prompt management, many children achieve long-term survival with good liver function
- Liver transplantation remains the definitive treatment for advanced disease, with excellent outcomes in Korean pediatric transplant centers